8-K: Current report filing
Published on August 15, 2014
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d)
of the Securities Exchange Act of 1934
Date of Report (Date of earliest event reported): August 15, 2014
NEOGENOMICS, INC.
(Exact name of registrant as specified in its charter)
| Nevada | 001-35756 | 74-2897368 | ||
| (State or other jurisdiction of incorporation) |
(Commission File Number) |
(I.R.S. Employer Identification No.) |
| 12701 Commonwealth Drive, Suite 9, Fort Myers, Florida |
33913 | |
| (Address of principal executive offices) | (Zip Code) |
(239) 768-0600
(Registrants telephone number, including area code)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
| ¨ | Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| ¨ | Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| ¨ | Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| ¨ | Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
FORWARD-LOOKING STATEMENTS
This Current Report on Form 8-K, contains forward-looking statements and information within the meaning of Section 27A of the Securities Act of 1933, as amended (the Securities Act), and Section 21E of the Securities Exchange Act of 1934, as amended (the Exchange Act), which are subject to the safe harbor created by those sections. These forward-looking statements include, but are not limited to, statements concerning our strategy, future operations, future financial position, future revenues, projected costs, prospects and plans and objectives of management. The words anticipates, believes, estimates, expects, intends, may, plans, projects, will, would and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements and you should not place undue reliance on our forward-looking statements. These forward-looking statements involve known and unknown risks and uncertainties that could cause our actual results, performance or achievements to differ materially from those expressed or implied by the forward-looking statements, including, without limitation, the risks set forth from time to time in the Companys filings with the SEC. Readers of this release are cautioned not to place undue reliance on forward-looking statements contained herein, which speak only as of the date stated, or if no date is stated, as of the date of this Current Report. The Company undertakes no obligation to publicly update or revise the forward-looking statements contained herein to reflect changes events or circumstances after the date of this release, unless required by law.
| Item 1.01 | Entry into a Material Definitive Agreement. |
On August 15, 2014, NeoGenomics, Inc., a Nevada corporation (the Company), entered into an underwriting agreement (the Underwriting Agreement) with William Blair & Company, L.L.C. (the Representative), acting as representative for the underwriters named in Schedule I of the Underwriting Agreement (the Underwriters), whereby the Company agreed to sell to the Underwriters, and the Underwriters agreed to purchase from the Company for resale to the public (the Offering), subject to the terms and conditions expressed therein, an aggregate of 7,000,000 shares (the Firm Securities) and, at the election of the Underwriters, up to 1,050,000 additional shares (the Optional Shares) of common stock, par value $0.001 per share (the Shares and each, a Share), of the Company (the Firm Securities and the Optional Shares are herein collectively referred to as the Securities). The price to the public will be $4.60 per Share. The option to purchase the Optional Shares is exercisable no later than 30 calendar days after the date of the Underwriting Agreement.
The Underwriting Agreement provides that the Company will indemnify the Underwriter against certain liabilities, including liabilities under the Securities Act of 1933, as amended, or to reimburse the Underwriter for payments that the Underwriter may be required to make because of such liabilities.
The Securities are being offered and sold pursuant to a prospectus supplement dated August 15, 2014 and an accompanying base prospectus, pursuant to the Companys existing shelf registration statement on Form S-3 (File No. 333-193105) that was declared effective by the Securities and Exchange Commission on January 3, 2014. The opinion of the Companys counsel regarding the validity of the Shares to be issued by the Company is filed herewith as Exhibit 5.1.
The foregoing description of the Underwriting Agreement is not complete and is qualified in its entirety by reference to the full text of the form of Underwriting Agreement, a copy of which is filed as Exhibit 1.1 to this Current Report on Form 8-K.
| Item 7.01. | Regulation FD. |
On August 15, 2014, the Company issued a press release announcing the pricing of the Offering (the Press Release). A copy of the Press Release is attached hereto as Exhibit 99.1, and is incorporated by reference herein.
This Current Report on Form 8-K shall not constitute an offer to sell or the solicitation of an offer to buy nor shall there be any sale of these Shares in any state in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any state. Any offering will be made only through a prospectus supplement and accompanying prospectus.
| Item 8.01 | Other Events. |
Updated Company Disclosure
The Company is filing the following information for the purpose of updating certain risk factors and information in connection with the Company and its business contained in its other filings with the Securities and Exchange Commission (SEC):
Risk Factors
Risks Related to Our Business
We May Be Unsuccessful In Managing Our Growth Which Could Prevent The Company From Operating Profitably
Our growth has placed, and is expected to continue to place, a significant strain on our managerial, operational and financial resources. To manage our potential growth, we must continue to implement and improve our operational, financial and billing systems and to expand, train and manage our employee base. We may not be able to effectively manage the expansion of our operations and our systems and our procedures or controls may not be adequate to support our operations. Our management may not be able to achieve the rapid execution necessary to fully exploit the market opportunity for our products and services. Any inability to manage growth could have a material adverse effect on our business, results of operations, potential profitability and financial condition. Part of our business strategy may be to acquire assets or other companies that will complement our existing business. At this time, we are unable to predict whether or when any material transaction will be completed should negotiations commence. If we proceed with any such transaction, we may not be able to effectively integrate the acquired operations with our own operations. We may also seek to finance any such acquisition by debt financings or issuances of equity securities and such financing may not be available on acceptable terms or at all.
The failure to obtain necessary additional capital to finance growth and capital requirements, could adversely affect our business, financial condition and results of operations.
We may seek to exploit business opportunities that require more capital than we have currently available. We may not be able to raise such capital on favorable terms or at all. If we are unable to obtain such additional capital, we may be required to reduce the scope of our anticipated expansion, which could adversely affect our business, financial condition and results of operations.
As of June 30, 2014, we had cash and cash equivalents of $5.0 million and had $8.0 million of availability under our credit facility with CapitalSource. We used approximately $3.0 million in cash and borrowed approximately $3.0 million on our credit facility, to finance the purchase of PathLogic on July 8, 2014.
Even if we are able to access the full amount available under our credit facility with CapitalSource, we may still need additional capital to fully implement our business, operating and development plans. Should the financing we require to sustain our working capital needs be unavailable or prohibitively expensive when we require it, there could be a material adverse effect on our long-term business, rate of growth, operating results, financial condition and prospects.
Proposed government regulation of laboratory developed tests may result in delays to launching certain laboratory tests and increase our costs to implement new tests.
We frequently develop testing procedures to provide diagnostic results to clients that cannot currently be provided using test kits approved by the U.S. Food and Drug Administration, or FDA. The FDA has been considering changes to the way that it regulates these Laboratory Developed Tests, or LDTs. Currently all LDTs are conducted and offered in accordance with Clinical Laboratory Improvements Amendments, or CLIAs, and individual state licensing procedures. The FDA is considering requiring FDA clearance or approval of a subset of LDTs, as well as a modified approach that may require FDA oversight short of the full approval process. There are
currently no formal definitions or regulations on how such approvals would be requested and granted, but there is a risk that such a process could delay the offering of certain tests and result in additional validation costs and fees. There is also an associated risk for us that some tests currently offered might become subject to the prior approval of the FDA. This FDA approval process would be time-consuming and costly, with no guarantee of ultimate approval success.
On July 31, 2014 the FDA issued a notification to Congress of the Anticipated Details of the Draft Guidance for Industry, Food and Drug Administration Staff, and Clinical Laboratories: Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs). As described in this notification, FDA plans to provide draft guidance to clinical laboratories that develop their own LDTs regarding how FDA intends to regulate such laboratories under the Federal Food, Drug, and Cosmetic Act. The anticipated regulatory framework would use a risk-based approach to enforce the FDAs premarket review requirements, and for high-risk tests, the framework may require laboratories to use FDA-approved tests, if available, rather than LDTs. If implemented, the framework may also require us to obtain premarket clearance or approval for certain of our LDTs. Implementation of this framework would include a lengthy phase-in period ranging from two to nine years depending on the risk assessment rating of each particular test. Once the draft guidance is issued, the FDA will provide an opportunity for public comment before the guidance is finalized. We anticipate the Agency will receive numerous comments on this issue, and the regulatory framework ultimately implemented by the FDA may differ substantially from the framework described in the notification to Congress.
Healthcare reform programs may impact our business and the pricing we receive for our services.
In March of 2010, health care reform legislation known as the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or commonly referred to collectively as the Affordable Care Act, was passed into law. The Affordable Care Act contains several provisions that seek to limit Medicare spending in the future. One key provision is the establishment of Accountable Care Organizations under which hospitals and physicians will be able to share savings that result from cost control efforts. We cannot predict what the final business models will be, nor can we predict with certainty the future impact on our business. There is the possibility that these organizations will seek to lower reimbursement for the services we provide and some may potentially restrict access to our services. We may not be able to gain access into certain Accountable Care Organizations. These changes could have an adverse and material impact on our operations. In furtherance of health care reform and the reduction in health care expenditures, the Affordable Care Act contains numerous provisions to be implemented through 2018. Other significant measures contained in the Affordable Care Act include, for example, coordination and promotion of research on comparative clinical effectiveness of different technologies and procedures, initiatives to revise Medicare payment methodologies, such as bundling of payments across the continuum of care by providers and physicians, and initiatives to promote quality indicators in payment methodologies. There can be no assurance at this time that the implementation of these provisions will not have a material adverse effect on our business.
In addition, other legislative changes have been proposed and adopted in the United States since the Affordable Care Act was enacted. On August 2, 2011, the Budget Control Act of 2011 among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislations automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers up to 2% per fiscal year, which went into effect on April 1, 2013 and will remain in effect through 2024 unless additional Congressional action is taken. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, or the ATRA, which, among other things, increased the period for the government to recover overpayments from providers from three to five years.
We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our services or additional pricing pressures.
Steps taken by government payers, such as Medicare and Medicaid to control the utilization and reimbursement of healthcare services, including esoteric testing may diminish our net revenue.
We face efforts by government payers to reduce utilization as well as reimbursement for laboratory testing services. Changes in governmental reimbursement may result from statutory and regulatory changes, retroactive rate adjustments, administrative rulings and other policy changes.
From time to time, legislative freezes and updates affect some of our tests that are reimbursed by the Medicare program under the Medicare Physician Fee Schedule or Clinical Laboratory Fee Schedule. The Medicare Physician Fee Schedule, which is updated on an annual basis using a prescribed statutory formula, is subject to significant reductions in reimbursement unless Congress intervenes. In the past, when the application of the statutory formula resulted in lower payments, Congress has passed interim legislation to prevent the reductions. The most recent legislative intervention passed was Protecting Access to Medicare Act of 2014, or PAMA, which provided for a 0.5% update from 2013 MPFS payment rates through 2014 and a 0% update from January 1 until April 1, 2015. If Congress fails to intervene to prevent the negative update factor in future years, the resulting decrease in payment may adversely affect our revenue, business, operating results, financial condition and prospects.
In addition, recent laws make changes to Medicare reimbursement for our tests that are reimbursed under the Clinical Laboratory Fee Schedule, or CLFS, many of which have already gone into effect. The Affordable Care Act includes a reduction in the annual update factor used to adjust payments under the CLFS for inflation. This update factor reflects the consumer price index for all urban consumers, or CPI-U, and the ACA reduces the CPI-U by 1.75% for the years 2011 through 2015. The Affordable Care Act also imposes a multifactor productivity adjustment in addition to the CPI-U, which may further reduce payment rates. Further, in February 2012, the Middle Class Tax Relief and Job Creation Act of 2012 was passed, which, among other things, reduced the update to the CLFS by an additional 2% for CY 2013, and rebased payments at the reduced rate for subsequent years. Overall, when adding this 2% reduction to the Affordable Care Acts adjustments, the payment rates under the CLFS declined by 2.95% and 0.75% for 2013 and 2014, respectively. This reduction does not include the additional sequestration adjustment.
Most recently, on April 1, 2014, PAMA was signed to law, which, among other things, is expected to significantly alter the current payment methodology under the CLFS. Under the new law, starting January 1, 2016 and every three years thereafter (or annually in the case of advanced diagnostic lab tests), clinical laboratories must report laboratory test payment data for each Medicare-covered clinical diagnostic lab test that it furnishes during a time period to be defined by future regulations. The reported data must include the payment rate (reflecting all discounts, rebates, coupons and other price concessions) and the volume of each test that was paid by each private payer (including health insurance issuers, group health plans, Medicare Advantage plans and Medicaid managed care organizations). Beginning in 2017, the Medicare payment rate for each clinical diagnostic lab test will be equal to the weighted median amount for the test from the most recent data collection period. The payment rate will apply to laboratory tests furnished by a hospital laboratory if the test is separately paid under the hospital outpatient prospective payment system. It is too early to predict the impact on reimbursement for our tests reimbursed under the CLFS.
Also under PAMA, the Centers for Medicare & Medicaid Services, or CMS, is required to adopt temporary billing codes to identify new tests and new advanced diagnostic laboratory tests that have been cleared or approved by the FDA. For an existing test that is cleared or approved by the FDA and for which Medicare payment is made as of April 1, 2014, CMS is required to assign a unique billing code if one has not already been assigned by the agency. In addition to assigning the code, CMS must publicly report payment for the tests no later than January 1, 2016. We cannot determine at this time the full impact of the new law on our business, financial condition and results of operations.
CMS also adopts policies, from time to time, limiting or excluding coverage for certain of the tests that we perform. Likewise, many state governments are under budget pressures and are also considering reductions to their Medicaid fees. Further, Medicare, Medicaid and other third party payers audit for overutilization of billed services. Even though all tests performed by us are ordered by our clients, who are responsible for establishing the medical necessity for the tests ordered, we may be subject to recoupment of payments, as the recipient of the payments for such tests, in the event that a third party payer such as CMS determines that the tests failed to meet all applicable criteria for payment. When third party payers like CMS revise their coverage policies, our costs generally increase
due to the complexity of complying with additional administrative requirements. Furthermore, Medicaid reimbursement and regulations vary by state. Accordingly, we are subject to varying administrative and billing regulations, which also increase the complexity of servicing such programs and our administrative costs. Finally, state budget pressures have encouraged states to consider several courses that may impact our business, such as delaying payments, restricting coverage eligibility, service coverage restrictions and imposing taxes on our services.
In certain jurisdictions, Palmetto GBA, a Medicare administrative contractor, administers the Molecular Diagnostic Services Program, or MolDX, and establishes coverage and reimbursement for certain molecular diagnostic tests, including many of our tests. To obtain coverage for an established molecular diagnostic test or LDT, laboratories must apply for and obtain a unique test identifier. For newly developed tests or for established tests that have not been validated for clinical and analytical validity and clinical utility, laboratories must submit a detailed dossier of clinical data to substantiate that the test meets Medicares requirements for coverage. We have received favorable coverage for many of our molecular tests, however we have also received non-coverage determination for many newer tests. The field of molecular diagnostics is evolving very rapidly, and clinical studies on many new tests are still underway. We cannot be assured that some of our molecular tests will ever be covered services by Medicare, nor can we determine when the medical literature will meet the standard for coverage that Palmetto GBA has set.
In recent years, Medicare has encouraged beneficiaries to participate in managed care programs, known as Medicare Advantage programs, and has encouraged beneficiaries from the traditional fee-for- service Medicare program to switch to Medicare Advantage programs. This has resulted in rapid growth of health insurance and managed care plans offering Medicare Advantage programs and growth in Medicare beneficiary enrollment in these programs. Also in recent years, many states have increasingly mandated that Medicaid beneficiaries enroll in managed care arrangements. If these efforts continue to be successful, we may experience a further shift of traditional Medicare and Medicaid fee-for-service beneficiaries to managed care programs. As a result, we would be required to contract with those private managed care programs in order to be reimbursed for services to their Medicare and Medicaid members. There can be no assurance that we will be successful in entering into agreements with these managed care programs at rates of payment similar to those we realize from our non-managed care lines of business.
CMS has, as part of its regulatory structure, developed the National Correct Coding Initiative, or NCCI to promote national correct coding methodologies and to control improper coding leading to inappropriate payment in Medicare Part B claims. The most recent NCCI Coding Policy Manual resulted in changes in how we bill both FISH and immunohistochemistry testing. The language relates to what NCCI considers bundled services, and will impact the quantity of certain tests that are billed. NCCI limits the number of units we may bill for certain test codes which lowers the overall reimbursement we receive for that test. While many in the laboratory industry are not in agreement with the determination, there can be no assurance that CMS will make any modifications to the existing language.
We expect the initiatives described above to continue and, if they do, to reduce reimbursements for clinical laboratory services, to impose more stringent cost controls on clinical laboratory services and to reduce utilization of clinical laboratory services. These efforts, including changes in law or regulations that may occur in the future, may each individually or collectively have a material adverse impact on our business, operating results, financial condition and prospects.
Third party billing is extremely complicated and results in significant additional costs to us.
Billing for laboratory services is extremely complicated. Depending on the billing arrangement and applicable law, we must bill various payers, such as patients, insurance companies, Medicare, Medicaid, doctors and employer groups, hospitals and other laboratories, all of which have different billing requirements. Additionally, our billing relationships require us to undertake internal audits to evaluate compliance with applicable laws and regulations as well as internal compliance policies and procedures. Insurance companies also impose routine external audits to evaluate payments made, which adds further complexity to the billing process.
Among others, the primary factors which complicate our billing practices are:
| | pricing differences between our fee schedules and the reimbursement rates of the payers; |
| | changes in carrier rules; |
| | disputes with payers as to the party who is responsible for payment; and |
| | disparity in coverage and documentation requirements among various carriers. |
We incur significant additional costs as a result of our participation in the Medicare and Medicaid programs, as billing and reimbursement for clinical laboratory services are subject to considerable and complex federal and state regulations. The additional costs we expect to incur include those related to: (i) complexity added to our billing processes and systems; (ii) training and education of our employees and clients; (iii) implementing compliance procedures and oversight; (iv) collections and legal costs; and (v) costs associated with, among other factors, challenging coverage and payment denials and providing patients with information regarding claims processing and services, such as advance beneficiary notices.
Our operations are subject to strict laws prohibiting fraudulent billing and other abuse, and our failure to comply with such laws could result in substantial penalties.
Of particular importance to our operations are federal and state laws prohibiting fraudulent billing and providing for the recovery of non-fraudulent overpayments. Government investigations of clinical laboratories have been ongoing for a number of years and are expected to continue in the future. A large number of laboratories have entered into substantial settlements with federal and state governments under these laws. Private payers have also brought civil actions against laboratories which have resulted in substantial judgments.
In particular, if an entity is determined to have violated the federal False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties of between $5,500 to $11,000 for each separate false claim. There are many potential bases for liability under the federal False Claims Act. Liability arises, when an entity submits, or causes another to submit, a claim for reimbursement to the federal government for a service which was not provided or which did not qualify for reimbursement. Submitting a claim with reckless disregard or deliberate ignorance of its truth or falsity could also result in substantial civil liability. In addition, the False Claims Acts whistleblower or qui tam provisions are being used with more frequency to challenge the reimbursement practices of providers and suppliers. Those provisions allow a private individual to bring an action on behalf of the government alleging that the defendant has submitted false claims for payment to the federal government. The government must decide whether to intervene in the lawsuit and whether to prosecute the case. If it declines to do so, the individual may pursue the case alone, although the government must be kept apprised of the progress of the lawsuit. Whether or not the federal government intervenes in the case, it will receive the majority of any recovery. The successful qui tam relator who brought the case is entitled to a portion of the proceeds and its attorneys fees and costs. In addition, various states have enacted laws modeled after the federal False Claims Act , which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer. If the government or qui tam relator were to allege or determine that we were violating these false claims laws, we could be subject to substantial fines and penalties, which could adversely affect our operations.
Risks Related to Our Common Stock
Actual operating results may differ significantly from our guidance.
From time to time, we release guidance regarding our future performance or the expected future performance of companies or businesses that we have agreed to acquire. Any such guidance represents our managements estimates as of the date of release. This guidance, which consists of forward-looking statements, is prepared by our management and is qualified by, and subject to, the assumptions and the other information contained or referred to in such release and the factors described under Forward-Looking Statements in this prospectus supplement. Our guidance is not prepared with a view toward compliance with published guidelines of the American Institute of Certified Public Accountants, and neither our independent registered public accounting firms nor any other independent expert or outside party compiles or examines the guidance and, accordingly, no such person expresses any opinion or any other form of assurance with respect thereto.
Guidance is based upon a number of assumptions and estimates that, while presented with numerical specificity, are inherently subject to business, economic and competitive uncertainties and contingencies, many of which are beyond our control and are based upon specific assumptions with respect to future business decisions, some of which will change. We generally state possible outcomes as high and low ranges which are intended to provide a sensitivity analysis as variables are changed but are not intended to represent that actual results could not fall outside of the suggested ranges. The principal reason that we release this data is to provide a basis for our management to discuss our business outlook with analysts and investors. We do not accept any responsibility for any projections or reports published by any such persons.
Guidance is necessarily speculative in nature, and it can be expected that some or all of the assumptions of the guidance furnished by us will not materialize or will vary significantly from actual results. Accordingly, our guidance is only an estimate of what management believes is realizable as of the date of release. Actual results will vary from the guidance. Investors should also recognize that the reliability of any forecasted financial data diminishes the farther in the future that the data is forecast. In light of the foregoing, investors are urged to put the guidance in context and not to place undue reliance on it. Any failure to successfully implement our operating strategy or the occurrence of any of the events or circumstances set forth in, or incorporated by reference into, this prospectus supplement could result in the actual operating results being different than the guidance, and such differences may be adverse and material.
Selected Financial Data
The charts set forth below provide selected financial and other annual and quarterly data which reflect results obtained by the Company during the periods covered by each respective chart. The results reflected in the charts below are not necessarily indicative of the results that should be expected in the future. Furthermore, interim results are not necessarily indicative of the results that should be expected for the full year or any other period.
Adjusted EBITDA is defined by NeoGenomics as net income from continuing operations before (i) interest expense, (ii) tax expense and therapeutic discovery tax grants, (iii) depreciation and amortization expense, (iv) non-cash stock-based compensation and warrant amortization expense and (v) other extraordinary or non-recurring charges, such as the costs related to moving our California facility. NeoGenomics believes that Adjusted EBITDA provides a more consistent measurement of operating performance and trends across reporting periods by excluding these cash and non-cash items of expense not directly related to ongoing operations from income. Adjusted EBITDA also assists investors in performing analysis that is consistent with financial models developed by research analysts.
Adjusted EBITDA as defined by NeoGenomics is not a measurement under GAAP and may differ from non-GAAP measures used by other companies. There are limitations inherent in non-GAAP financial measures such as Adjusted EBITDA because they exclude a variety of charges and credits that are required to be included in a GAAP presentation, and do not therefore present the full measure of NeoGenomics recorded costs against its net revenue. Accordingly, investors should consider non-GAAP results together with GAAP results in analyzing NeoGenomics financial performance.
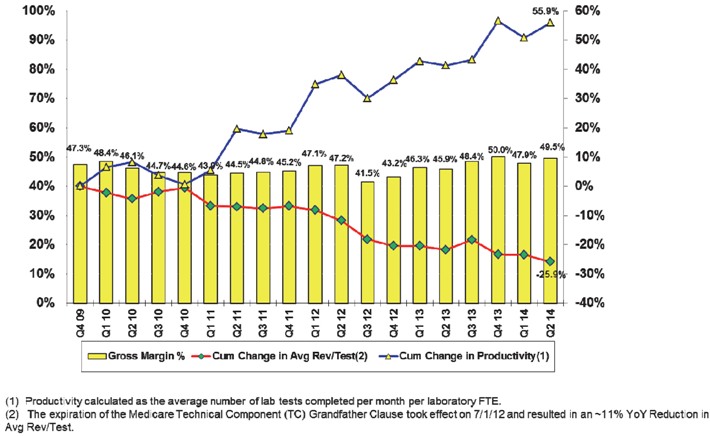
The chart below provides additional information regarding our historical gross margin, cumulative change in average revenue per test and cumulative change in productivity. In footnote (1), FTE means full time equivalent, and in footnote (2), YoY means year-over-year.
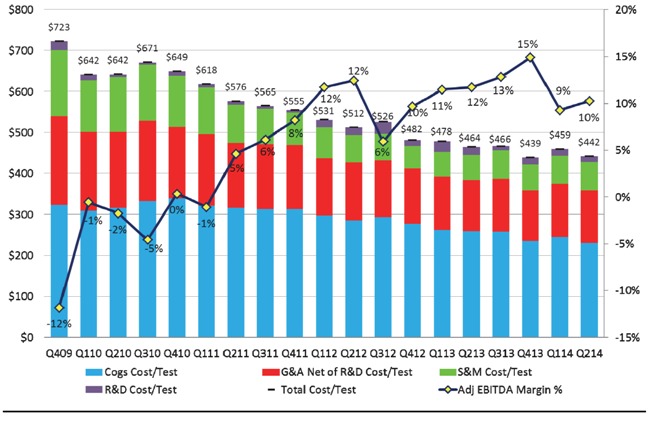
The chart below provides additional information regarding our historic cost per test and adjusted EBITDA margin percentage.
| Item 9.01. | Financial Statements and Exhibits. |
(d) Exhibits
| 1.1 | Form of Underwriting Agreement | |
| 5.1 | Opinion of Burton, Bartlett & Glogovac. | |
| 23.1 | Consent of Burton, Bartlett & Glogovac (included as part of Exhibit 5.1). | |
| 99.1 | Press Release dated August 15, 2014. | |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| NEOGENOMICS, INC. | ||
| By: | /s/ George A. Cardoza |
|
| George A. Cardoza | ||
| Chief Financial Officer | ||
Date: August 15, 2014